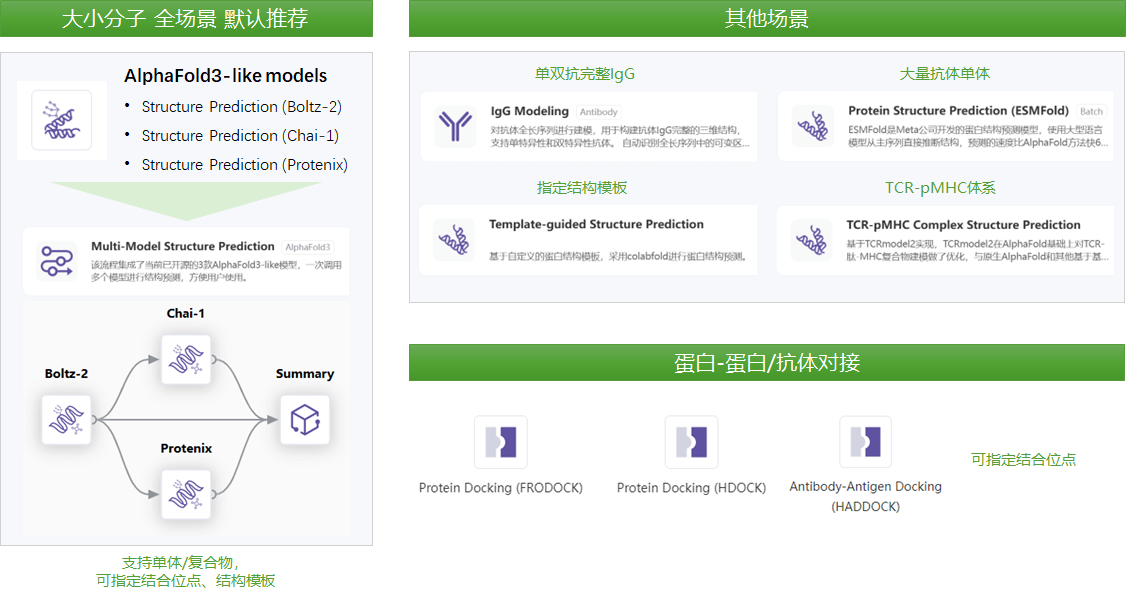
请根据需要预测的分子形式,选用对应的模块或流程,如图所示:
比较常用的模块推荐如下:
| 模块、流程 | 适用场景/体系 |
|---|---|
| Multi-model Structure Prediction | 大部分场景首选(多数情况默认推荐):抗体可变区结构预测,抗体-抗原复合物结构预测,多肽,蛋白-小分子复合物,酶-底物,核酸等 |
| Structure Prediction (Protenix):Enhanced Mode(增强采样模式) | 对精度要求高的场景, 在Multi-model Structure Prediction 的基础上,探索更多的可能性 |
| Protein Structure Prediction (ESMFold) | 快速、批量的抗体可变区结构预测,主要用于对精度要求不高的计算或初步筛选,例如人源化设计、表达量排序 |
| IgG Modeling | 完整抗体IgG结构预测、对称式IgG+scFv/VHH |
以下是部分功能模块的说明:
| 模块/流程 | 描述 |
|---|---|
| Multi-Model Structure Prediction | 一次调用多个AlphaFold3-like模型进行结构预测 |
| Structure Prediction (Boltz-2) | AlphaFold3-like结构预测模型,基于MIT的Boltz-2模型 |
| Structure Prediction (Protenix) | AlphaFold3-like结构预测模型,基于字节的Protenix模型 |
| Structure Prediction (Chai-1) | AlphaFold3-like结构预测模型,基于Chai Discovery的Chai-1模型 |
| Protein Structure Prediction (ESMFold) | 速度快,可用于大量抗体可变区单体结构预测 |
| Immune Protein Structure Prediction | 基于ImmuneBuilder的免疫蛋白结构预测,包括单抗(ABodyBuilder2)、VHH(NanoBodyBuilder2)、TCR(TCRBuilder2)等的可变区预测 |
| IgG Modeling | 完整抗体IgG结构预测,目前支持常规单抗、不对称IgG双抗,以及对称式IgG+scFv/VHH(通过WeFormat双抗编辑器输入),暂不支持其他多特异性format |
| Cyclic Peptide Structure Prediction | 环肽结构预测 |
| RNA Secondary Structure Prediction | RNA二级结构预测 |
| RNA 3D Structure Prediction | RNA三级结构预测 |
| Complex Structure Prediction & PPI Scoring | 一次调用多个AlphaFold3-like模型进行结构预测,同时使用PPI亲和力预测模块对预测结构进行能量评估,辅助复合物结构的挑选 |
Please select the corresponding module or workflow based on the molecular format you need to predict, as shown in the figure:
The most commonly used module recommendations are as follows:
| Module/Workflow | Applicable Scenarios/Systems |
|---|---|
| Multi-model Structure Prediction | First choice for most scenarios (default recommendation in most cases): antibody variable region structure prediction, antibody-antigen complex structure prediction, peptides, protein-small molecule complexes, enzyme-substrate, nucleic acids, etc. |
| Structure Prediction (Protenix): Enhanced Mode | For scenarios requiring high accuracy: explores more possibilities based on Multi-model Structure Prediction |
| Protein Structure Prediction (ESMFold) | Fast, batch antibody variable region structure prediction, mainly used for calculations or preliminary screening that do not require high accuracy, such as humanization design and expression level ranking |
| IgG Modeling | Full antibody IgG structure prediction, symmetric IgG+scFv/VHH |
The following are descriptions of some functional modules:
| Module/Workflow | Description |
|---|---|
| Multi-Model Structure Prediction | Invokes multiple AlphaFold3-like models simultaneously for structure prediction |
| Structure Prediction (Boltz-2) | AlphaFold3-like structure prediction model, based on MIT’s Boltz-2 model |
| Structure Prediction (Protenix) | AlphaFold3-like structure prediction model, based on ByteDance’s Protenix model |
| Structure Prediction (Chai-1) | AlphaFold3-like structure prediction model, based on Chai Discovery’s Chai-1 model |
| Protein Structure Prediction (ESMFold) | Fast speed, suitable for large-scale antibody variable region monomer structure prediction |
| Immune Protein Structure Prediction | Immune protein structure prediction based on ImmuneBuilder, including variable region prediction for monoclonal antibodies (ABodyBuilder2), VHH (NanoBodyBuilder2), TCR (TCRBuilder2), etc. |
| IgG Modeling | Full antibody IgG structure prediction, currently supports conventional monoclonal antibodies, asymmetric IgG bispecific antibodies, and symmetric IgG+scFv/VHH (input via WeFormat bispecific editor), other multispecific formats are not yet supported |
| Cyclic Peptide Structure Prediction | Cyclic peptide structure prediction |
| RNA Secondary Structure Prediction | RNA secondary structure prediction |
| RNA 3D Structure Prediction | RNA 3D structure prediction |
| Complex Structure Prediction & PPI Scoring | Invokes multiple AlphaFold3-like models simultaneously for structure prediction, while using the PPI affinity prediction module to evaluate the energy of predicted structures, assisting in the selection of complex structures |
对PDB结构文件进行处理,包括去除杂质、补全确实原子或残基、加氢、修改链名或残基编号等。
| 模块/流程名称 | 描述 |
|---|---|
| Structure Preparation | 首选,支持提取链,去除杂质,补全缺失原子、残基,以及蛋白氨基酸残基的质子化判断以及加氢等操作 |
| Structure Minimization | 结构优化模块,支持氢原子优化、氨基酸侧链优化、整体优化三种方式 |
| PDB ReNumbering | 针对蛋白PDB文件中残基重新编号的工具模块,指定残基开始编号序号,同时支持抗体kabat,imgt以及chothia的重编号 |
| PDB Mutation | 用于突变PDB格式的蛋白质结构并返回突变后的结构 |
Processing of PDB structure files includes removing impurities, completing missing atoms or residues, adding hydrogen atoms, modifying chain names or residue numbers, etc.
| Module/Process Name | Description |
|---|---|
| Structure Preparation | Preferred option, supports chain extraction, impurity removal, completion of missing atoms and residues, and operations like protonation judgment and hydrogen addition for protein amino acid residues |
| Structure Minimization | Structure optimization module, supports three methods: hydrogen atom optimization, amino acid side chain optimization, and overall optimization |
| PDB ReNumbering | Tool module for renumbering residues in protein PDB files, specifying the starting number for residues, and supporting renumbering according to Kabat, IMGT, and Chothia schemes |
| PDB Mutation | Used for mutating protein structures in PDB format and returning the mutated structure |
Introduction to Simulated Structure Processing Documentation
RMSD (Root Mean Square Deviation) 和 DockQ 都是评估分子结构相似性和对接模型质量的指标,但它们的应用范围和考量因素有所不同。
定义: RMSD 是衡量两个叠加的分子结构之间原子位置平均偏差的量度。它通过计算对应原子(通常是主链原子,如 Cα 原子,或所有重原子)在三维空间中的距离平方的均值,再开平方根得到。
应用场景:
特点:
定义: DockQ 是一个专门用于评估蛋白质-蛋白质对接模型质量的连续性指标,范围在 [0,1] 之间。它结合了多个衡量对接质量的关键因素,以提供一个更全面、更接近 CAPRI (Critical Assessment of PRediction of Interactions) 评估标准的单一分数。
组成部分: DockQ 综合了以下几个关键指标:
计算方式: DockQ 并非简单的线性组合,而是通过对这些组分进行非线性变换和组合得出的,旨在更好地重现 CAPRI 的质量分类(Incorrect, Acceptable, Medium, High)。
应用场景:
特点:
| 特征 | RMSD (Root Mean Square Deviation) | DockQ |
|---|---|---|
| 应用范围 | 广泛用于各种分子结构比较(蛋白质、小分子、构象变化) | 主要用于蛋白质-蛋白质对接模型质量评估 |
| 评估目标 | 衡量两个结构之间原子位置的几何相似性 | 衡量蛋白质-蛋白质对接模型在界面区域的准确性 |
| 考量因素 | 仅考虑原子位置的几何偏差 | 综合考虑界面 RMSD、配体 RMSD 和天然接触分数 |
| 结果形式 | 距离单位(Å),越小越好 | 0-1 之间的连续分数,越大越好 |
| 侧重点 | 全局或局部结构相似性 | 蛋白质相互作用界面的准确性和生物学相关性 |
| 与对接关系 | 可以作为对接评估的一个组成部分(如iRMSD, LRMSD) | 专门为蛋白质对接设计,整合了多个对接相关指标 |
简而言之,RMSD 是一个更通用的几何相似性度量,可以用于各种分子结构比较。而 DockQ 则是一个专门为蛋白质-蛋白质对接模型设计的高度集成的质量评估指标,它更全面地反映了对接的生物学相关性和准确性,因为它综合了界面几何精度和关键相互作用的正确性。在评估蛋白质-蛋白质对接时,DockQ 通常被认为是更优选和更具代表性的指标。
RMSD (Root Mean Square Deviation) and DockQ are both metrics used to evaluate molecular structure similarity and docking model quality, but they differ in their range of applications and considerations.
Definition: RMSD is a measure of the average deviation in atomic positions between two superimposed molecular structures. It is calculated by taking the square root of the mean of the squared distances between corresponding atoms (typically backbone atoms, such as Cα atoms, or all heavy atoms) in three-dimensional space.
Applications:
Characteristics:
Definition: DockQ is a continuous metric specifically designed to evaluate the quality of protein-protein docking models, ranging from [0,1]. It combines multiple key factors for assessing docking quality to provide a more comprehensive score that aligns closely with CAPRI (Critical Assessment of PRediction of Interactions) evaluation standards.
Components: DockQ integrates the following key metrics:
Calculation Method: DockQ is not a simple linear combination but is derived through nonlinear transformations and combinations of these components, aiming to better reproduce CAPRI’s quality classifications (Incorrect, Acceptable, Medium, High).
Applications:
Characteristics:
| Feature | RMSD (Root Mean Square Deviation) | DockQ |
|---|---|---|
| Scope of Application | Widely used for various molecular structure comparisons (proteins, small molecules, conformational changes) | Primarily used for evaluating the quality of protein-protein docking models |
| Evaluation Target | Measures geometric similarity of atomic positions between two structures | Measures the accuracy of the interface region in protein-protein docking models |
| Considered Factors | Considers only geometric deviations of atomic positions | Integrates interface RMSD, ligand RMSD, and fraction of native contacts |
| Result Format | Distance unit (Å), smaller is better | Continuous score between 0-1, higher is better |
| Focus | Global or local structural similarity | Accuracy and biological relevance of protein interaction interfaces |
| Relation to Docking | Can be a component of docking evaluation (e.g., iRMSD, LRMSD) | Specifically designed for protein docking, integrating multiple docking-related metrics |
In short, RMSD is a more general metric for geometric similarity, applicable to various molecular structure comparisons. DockQ, on the other hand, is a highly integrated quality assessment metric specifically designed for protein-protein docking models, providing a more comprehensive reflection of the biological relevance and accuracy of docking by integrating interface geometric precision and the correctness of key interactions. In evaluating protein-protein docking, DockQ is often considered a more preferred and representative metric.
对于入门用户,可使用
对于有经验的用户,建议使用WeSeq中的交互式人源化设计面板:WeSeq->Humanization,可以基于人工经验调整人源化设计方案,实时对照结构进行更精细化的设计。
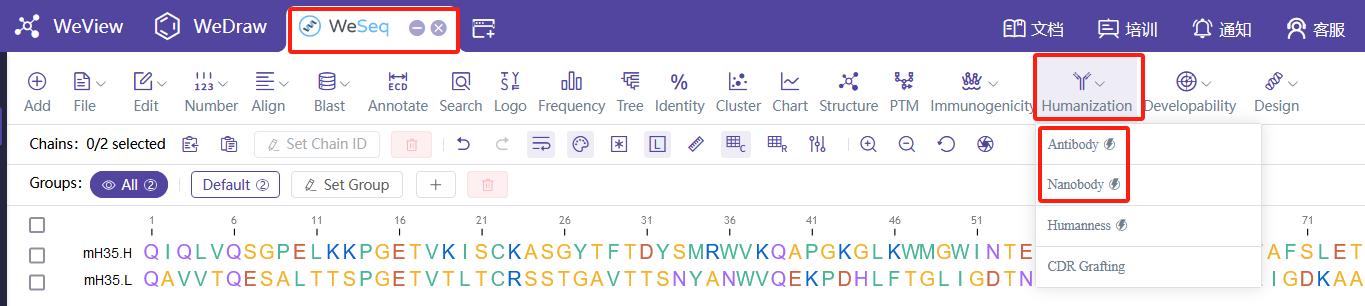
For beginner users, the following can be used:
Humanization Process Introduction Document
For experienced users, it is recommended to use the interactive humanization design panel in WeSeq: WeSeq->Humanization, which allows for manual adjustment of the humanization design scheme based on experience and real-time comparison with the structure for more refined design.
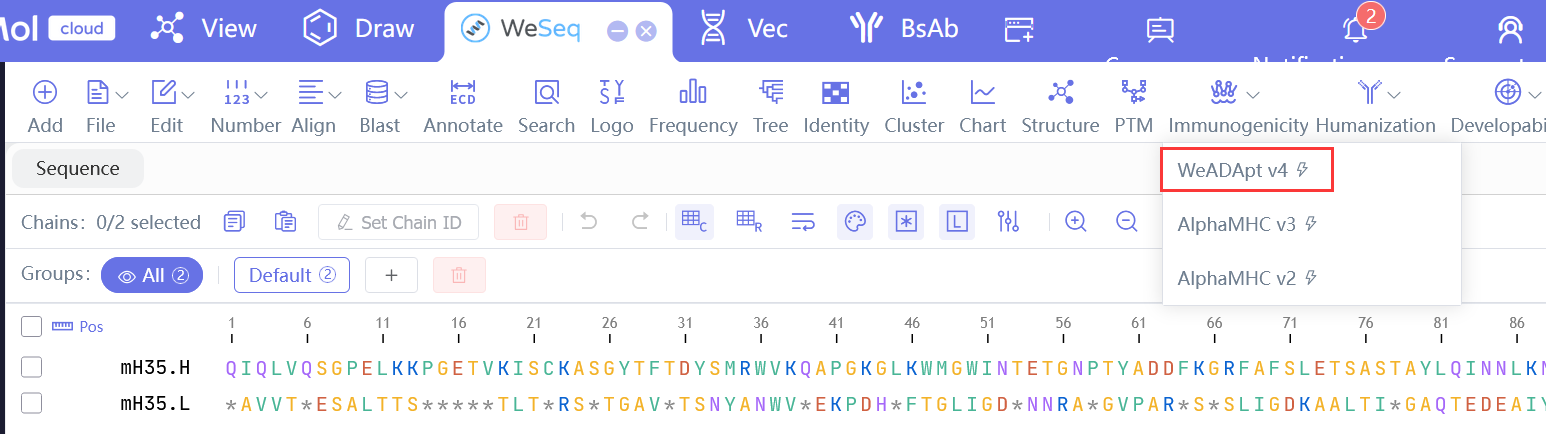
免疫原性预测已经历多个版本迭代,目前推荐版本为:WeADApt v4.3,WeADApt v4.2, AlphaMHC v3.0 beta。
同时也可以从WeSeq中提交预测:WeSeq->Immunogenicity,界面更友好(推荐v4)。
Immunogenicity prediction has undergone multiple version iterations. The currently recommended versions are: WeADApt v4.3, WeADApt v4.2, AlphaMHC v3.0 beta.
You can also submit predictions from WeSeq: WeSeq->Immunogenicity, which offers a more user-friendly interface (v4 recommended).
基于复合物结构的方法
针对复合物结构的相互作用界面进行饱和突变或进化优势突变,再用物理方法计算能量。建议使用亲和力成熟流程:
抗体亲和力成熟,使用Antibody Virtual Affinity Maturation流程。
蛋白亲和力成熟,使用Protein Virtual Affinity Maturation流程。
基于配体的方法
建议使用模块AA Probability Prediction,大语言模型预测高概率AA。主要是利用大语言模型和配体的序列(或结构)直接推荐高适应性(fitness)突变。
蛋白复合物亲和力的相对结合自由能计算,可使用模块Protein FEP。
基于ESMIF逆折叠模型,预测能提升结构亲和力的单点或多点突变,可使用模块Structure Evolution。
For interaction interfaces of complex structures, perform saturation mutations or evolutionarily advantageous mutations, then use physical methods to calculate energy. It is recommended to use the affinity maturation workflows:
For antibody affinity maturation, use Antibody Virtual Affinity Maturation.
For protein affinity maturation, use Protein Virtual Affinity Maturation.
Affinity Maturation Introduction Document
Ligand-based methods It is recommended to use the module AA Probability Prediction, where large language models predict high-probability amino acids. This approach primarily leverages large language models along with the ligand’s sequence (or structure) to directly recommend high-fitness mutations.
For calculating the relative binding free energy of protein complex affinity, the module Protein FEP can be used.
Based on the ESM-IF inverse folding model, to predict single or multiple mutations that can enhance structural affinity, the module Structure Evolution can be used.
热稳定性与蛋白的折叠自由能正相关,可能影响表达、纯度、PK等,优化方式包括基于物理的能量计算和ML/AI模型。
优化抗体稳定性,可使用最新版Antibody Stability Optimization。
抗体稳定性优化流程介绍文档
优化蛋白稳定性,可使用最新版Protein Stability Optimization。
蛋白稳定性优化流程介绍文档
预测蛋白质的绝对稳定性,可使用Absolute Folding Stability。
蛋白绝对稳定性预测介绍文档
预测蛋白稳定性相对结合自由能,可使用Protein FEP。
基于ThermoMPNN模型预测蛋白质单点突变的稳定性变化,可使用Mutation Energy of Stability (ThermoMPNN)。
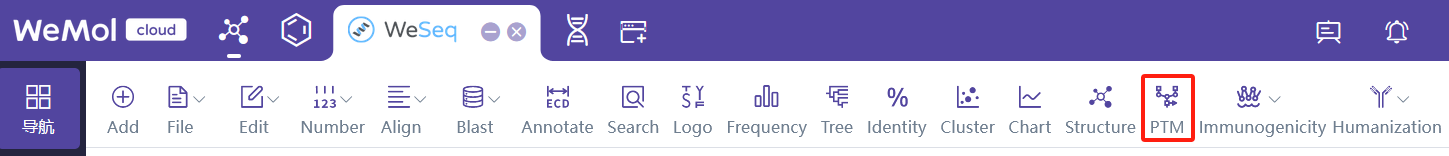
基于序列预测蛋白中潜在的PTM位点,可使用PTM Hotspot by Sequence。建议在WeSeq中进行分析:WeSeq->PTM。
基于结构预测蛋白中潜在的PTM位点,可使用PTM Hotspot by Structure。
基于ESMIF逆折叠模型,预测能提升结构稳定性的单点或多点突变,可使用Structure Evolution。
Thermal stability is positively correlated with the folding free energy of proteins, which may affect expression, purity, pharmacokinetics (PK), etc. Optimization methods include physics-based energy calculations and ML/AI models.
To optimize antibody stability, you can use Antibody Stability Optimization.
Antibody Stability Optimization Process Introduction Document
To optimize protein stability, you can use Protein Stability Optimization.
Protein Stability Optimization Process Introduction Document
To predict the absolute stability of proteins, you can use Absolute Folding Stability.
Absolute Folding Stability Prediction Introduction Document
To predict the relative binding free energy of protein stability, you can use Protein FEP.
To predict the stability changes of protein single-point mutations based on the ThermoMPNN model, you can use Mutation Energy of Stability (ThermoMPNN).
To predict potential PTM sites in proteins based on sequence, you can use PTM Hotspot by Sequence. It is recommended to perform the analysis in WeSeq: WeSeq -> PTM.
To predict potential PTM sites in proteins based on structure, you can use PTM Hotspot by Structure.
可开发性包括蛋白表面patch分析、理化性质计算(含pI)、TAP原则、PTM(基于序列)、基于结构的异构化预测、断裂位点预测等。
成药性一键综合评价
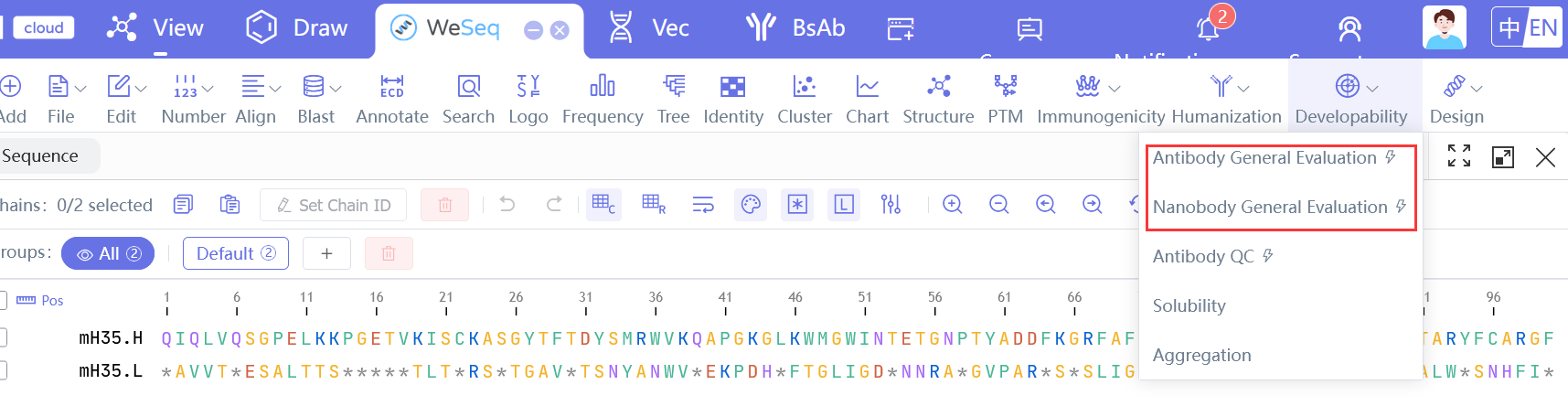
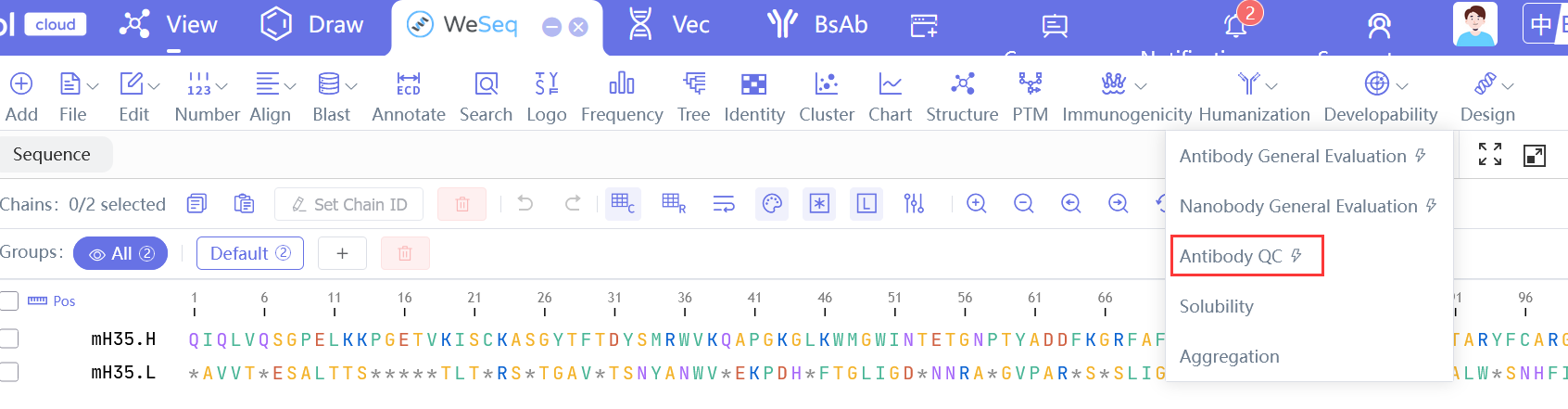
抗体QC流程
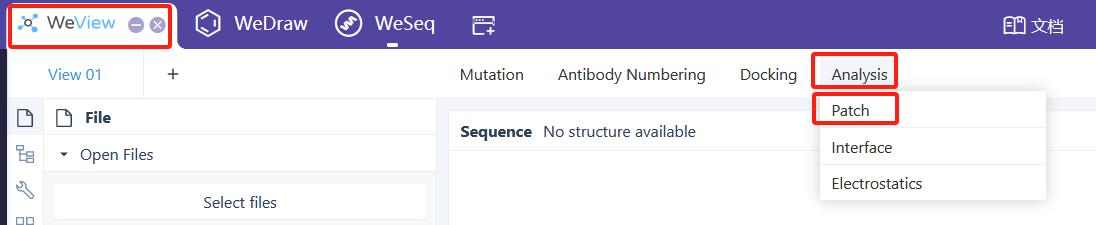
Patch分析
建议从WeView中运行:WeView->Analysis->Patch。Patch分析介绍文档
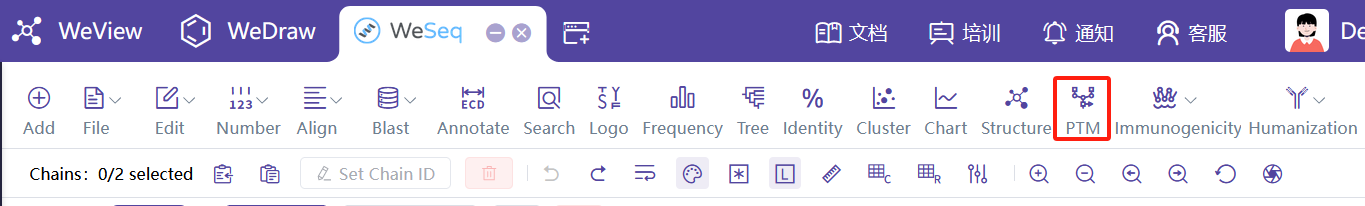
PTM预测
基于序列的PTM预测,建议直接在WeSeq运行:WeSeq->PTM。PTM预测介绍文档
基于结构的PTM预测,可以直接在模块中运行:PTM Hotspot by Structure。
抗体成药性预测(TAP)
溶解度预测
聚集度预测
Developability includes protein surface patch analysis, physicochemical property calculations (including pI), TAP principles, PTM (sequence-based), structure-based isomerization prediction, cleavage site prediction, etc.
One-click Comprehensive Druggability Assessment
Antibody QC Workflow
Patch Analysis
Recommended to run from WeView: WeView->Analysis->Patch. Patch Analysis Documentation
PTM Prediction
For sequence-based PTM prediction, it is recommended to run directly in WeSeq: WeSeq->PTM. PTM Prediction Documentation
For structure-based PTM prediction, you can run directly in the module: PTM Hotspot by Structure.
Antibody Druggability Prediction (TAP)
Solubility Prediction
Aggregation Prediction
序列分析包括序列编号、多序列比对、测序数据分析、频率分析、序列突变等。
序列编号
进行抗体序列编号,建议在WeSeq中运行:WeSeq->Number。序列编号介绍文档
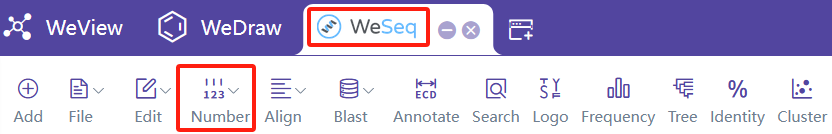
多序列比对
进行多序列比对,建议在WeSeq中运行:WeSeq->Align。多序列比对介绍文档

测序数据分析
进行测序数据分析,可以使用模块NGS Analysis。NGS Analysis介绍文档
频率分析
进行频率分析,建议在WeSeq运行,WeSeq->Frequency。频率分析介绍文档

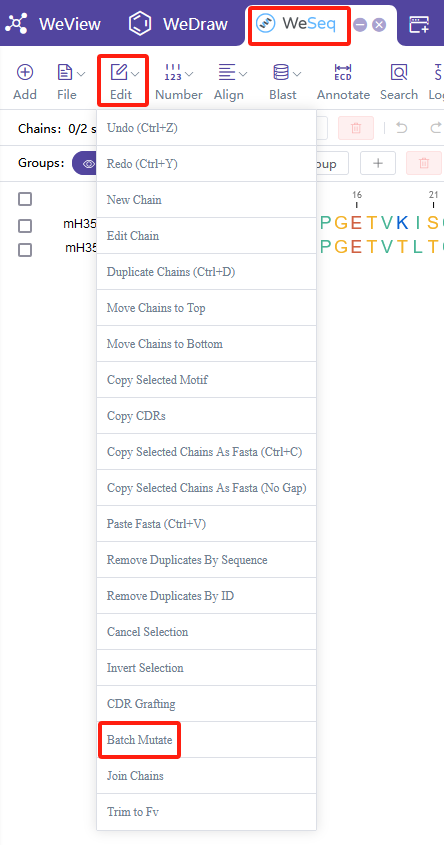
序列突变
进行序列突变,建议在WeSeq中操作:WeSeq->Edit->Batch Mutate。或者使用Sequence Mutation模块。
Sequence analysis includes sequence numbering, multiple sequence alignment, sequencing data analysis, frequency analysis, and sequence mutations.
For antibody sequence numbering, it is recommended to run in WeSeq: WeSeq -> Number. Sequence Numbering Introduction Document
For multiple sequence alignment, it is recommended to run in WeSeq: WeSeq -> Align. Multiple Sequence Alignment Introduction Document
For sequencing data analysis, you can use NGS Analysis. NGS Analysis Introduction Document
For frequency analysis, it is recommended to run in WeSeq: WeSeq -> Frequency. Frequency Analysis Introduction Document
For sequence mutation, it is recommended to operate in WeSeq: WeSeq -> Edit -> Batch Mutate. Alternatively, you can use the Sequence Mutation module.
专利分析包括专利抗体CDR序列搜索、专利序列提取、专利图片OCR。专利分析介绍文档
进行专利抗体CDR序列搜索,可以应用模块Patent BLAST。
从专利文本文件或专利序列图片OCR提取专利序列,可以应用模块Patent Sequence Listing。
Patent analysis includes searching for antibody CDR sequences in patents, extracting patent sequences, and performing OCR on patent images. Patent Analysis Introduction Document
To search for antibody CDR sequences in patents, you can use Patent CDR BLAST.
To extract sequences from patent text files or perform OCR on patent sequence images, you can use Patent Sequence Listing.
从头结构生成
进行蛋白结构从头生成,可以应用Protein Design (RFDiffusion)。RFDiffusion介绍文档
基于主链结构设计序列(逆折叠)
ProteinMPNN,建议从WeSeq中运行:WeSeq->Design->ProteinMPNN。ProteinMPNN介绍文档
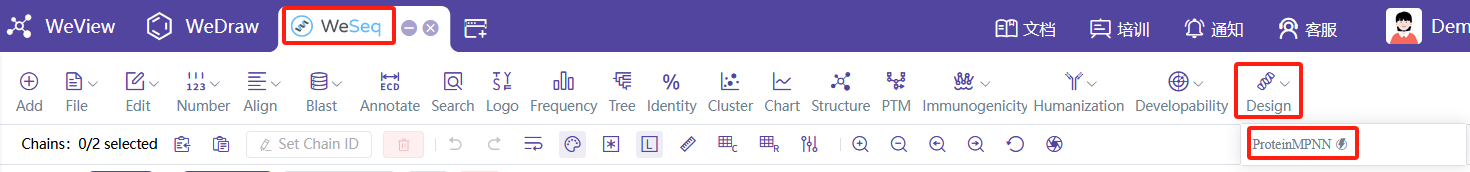
ABACUS-R模型,可以使用模块Protein Design (ABACUS-R)。
RFDesign模型,可以使用模块Protein Design (RFDesign)。RFDesign介绍文档。
ESMIF逆折叠模型,可使用模块Structure Evolution。
To perform de novo protein structure generation, you can use Protein Design (RFDiffusion). RFDiffusion Introduction Document
ProteinMPNN
ABACUS-R
RFDesign
ESMIF Inverse Folding Model
RFAntibody
是基于RFAntibody(抗体微调版RFdiffusion)的抗体从头设计。Antibody Design (RFAntibody)。
MEAN模型
基于MEAN模型实现的抗体设计,该模型采用多通道等变图注意力网络,可用于设计CDR的一维序列和三维结构。Antibody Design (MEAN)。
DiffAb模型
基于扩散概率模型和等价神经网络的抗体设计,可针对特定抗原结构生成抗体,也可基于抗体-抗原复合物结构进行抗体结构和序列的优化。Antibody Design (DiffAb)。
RFAntibody
RFAntibody is an antibody de novo design method based on the fine-tuned version of RFdiffusion. Antibody Design (RFAntibody).
MEAN Model
The MEAN model enables antibody design using a multi-channel equivariant graph attention network, which can be used to design both the one-dimensional sequence and three-dimensional structure of CDRs. Antibody Design (MEAN).
DiffAb Model
The DiffAb model utilizes diffusion probabilistic models and equivariant neural networks for antibody design. It can generate antibodies specific to a given antigen structure and optimize antibody structure and sequence based on antibody-antigen complex structures. Antibody Design (DiffAb).
多肽分析包括线性肽/环肽结构预测、多肽对接筛选、线性肽/环肽设计、信号肽预测。
Peptide analysis includes linear/cyclic peptide structure prediction, peptide docking screening, linear/cyclic peptide design, and signal peptide prediction.
包括密码子优化、CDS优化、UTR优化等。
Including codon optimization, CDS optimization, UTR optimization, etc.
靶点鉴定包括疾病相关靶点提取以及小分子靶点预测模块。靶点鉴定介绍文档
Target identification includes disease-related target extraction and small molecule target prediction modules. Target Identification Introduction Document
小分子生成是从头设计全新分子的过程,可以基于多种AI架构生成类药分子,也可以基于靶点,骨架、活性分子生成衍生物或者相似分子。分子生成介绍文档
Lead Optimization
先导化合物优化流程,包括基于阳性分子的结构生成、分子三维相似度筛选、分子对接以及ADMET预测,筛选获得优化分子
Small Molecule Generation (GenMol)
基于diffusion model的开源AI框架,用于分子生成。它从大型化学数据库中学习,生成类药物分子。GenMol能够同时优化多种属性(类药物特性、合成可得性),并提供合成规划,大致确保分子可在实验室中合成。
Small Molecule Generation (EvoMol)
智能分子生成功能,EvoMol支持全局结构优化与指定子结构的局部优化两种模式,能够在更短时间内批量产生多样性更高、结构新颖性更强的候选分子。
Small Molecule Generation (REINVENT4)
基于REINVENT4的小分子生成。支持多种分子生成方式:Reinvent - 从头开始创造新分子,Libinvent - 修饰一个骨架,Linkinvent - 设计两个片段之间的linker,Mol2Mol - 在用户定义的相似度范围内优化分子。
Scaffold Constrained Small Molecule Generation
为骨架限制的生成模型,可以限制骨架,指定优化部位,特异性的生成全新分子库。
Small Molecule Generation from Pocket
基于DiffSBDD模型实现,可应用受体的结合口袋生成小分子配体。
Small Molecule Random Generation
随机类药分子生成模型,基于多种主流的分子生成模型,包括字符级循环神经网络,变分自编码器,以及对抗自编码器的分子生成模块。
Small molecule generation is the process of de novo design of entirely new molecules. It can generate drug-like molecules based on various AI architectures, and can also generate derivatives or similar molecules based on targets, scaffolds, or active molecules. Molecular Generation Documentation
Lead Optimization
Lead compound optimization workflow, including structure generation based on positive molecules, molecular 3D similarity screening, molecular docking, and ADMET prediction to screen and obtain optimized molecules.
Small Molecule Generation (GenMol)
An open-source AI framework based on diffusion models for molecular generation. It learns from large chemical databases to generate drug-like molecules. GenMol can simultaneously optimize multiple properties (drug-likeness, synthetic accessibility) and provides synthetic planning to roughly ensure molecules can be synthesized in the laboratory.
Small Molecule Generation (EvoMol)
Intelligent molecular generation function. EvoMol supports two modes: global structure optimization and local optimization of specified substructures, capable of generating candidate molecules with higher diversity and stronger structural novelty in shorter time.
Small Molecule Generation (REINVENT4)
Small molecule generation based on REINVENT4. Supports multiple molecular generation methods: Reinvent - create new molecules from scratch, Libinvent - modify a scaffold, Linkinvent - design linkers between two fragments, Mol2Mol - optimize molecules within user-defined similarity ranges.
Scaffold Constrained Small Molecule Generation
A scaffold-constrained generation model that can restrict scaffolds, specify optimization sites, and specifically generate entirely new molecular libraries.
Small Molecule Generation from Pocket
Based on the DiffSBDD model, it can generate small molecule ligands using the receptor’s binding pocket.
Small Molecule Random Generation
Random drug-like molecule generation model based on various mainstream molecular generation models, including character-level recurrent neural networks, variational autoencoders, and adversarial autoencoder molecular generation modules.
虚拟筛选根据配体或受体结构,对小分子化合物进行筛选,预测可能的活性分子,大大提高化合物药物发现进程,缩减药物发现费用。
Virtual screening is a computational technique used to identify potential active compounds by screening large libraries of small molecules. This process can significantly accelerate drug discovery and reduce costs.
Property Filtering
Structure Search
3D Shape Search
Structure Clustering
分子性质包括小分子的理化性质以及药代动力学(ADMET)性质。
Molecular properties include the physicochemical properties and pharmacokinetic (ADMET) properties of small molecules.
分子对接是研究相互作用的重要工具,包括蛋白-小分子,蛋白-蛋白对接。
蛋白-小分子对接
Molecular Docking (AutoDock-GPU),建议从WeView中运行:WeView->Docking。基于GPU加速的AutoDock的分子对接工具。AutoDock-GPU对接介绍文档
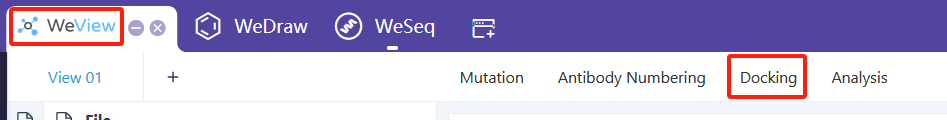
Molecular Docking (SMINA),基于Autodock Vina分支SMINA的分子对接工具。SMINA对接介绍文档
Molecular Docking (DiffDock),基于扩散生成模型的对接工具。DiffDock对接介绍文档
Molecular Docking (Gnina),基于深度学习的分子对接工具,采用卷积神经网络(CNN)评分函数对配体-受体结合构象进行打分和排序。
蛋白-蛋白/核酸对接
Molecular docking is an important tool for studying interactions, including protein-small molecule and protein-protein docking.
Protein-Small Molecule Docking
Molecular Docking (AutoDock-GPU), recommended to run from WeView: WeView->Docking. A GPU-accelerated molecular docking tool based on AutoDock. AutoDock-GPU Docking Documentation
Molecular Docking (SMINA), a molecular docking tool based on the AutoDock Vina branch SMINA. SMINA Docking Documentation
Molecular Docking (DiffDock), a docking tool based on diffusion generative models. DiffDock Docking Documentation
Molecular Docking (Gnina), A deep learning-based molecular docking tool that employs convolutional neural network (CNN) scoring functions to score and rank ligand-receptor binding poses.
Protein-Protein/Nucleic Acid Docking
分子格式转换工具,包括不同格式文件转换、氨基酸字母格式转换等。
Molecular format conversion tools, including conversion of different format files, amino acid letter format conversion, etc.
轨迹分析对分子动力学模拟后产生的轨迹进行结构分析,观察研究对象在模拟过程中的动态变化。
| 模块/流程名称 | 描述 |
|---|---|
| MD Trajectory | 可根据起始帧数、结束帧数以及间隔帧数对平衡模拟进行轨迹提取,并将其转换为GRO或者PDB格式文件 |
| MD RMS | 体系结构稳定性分析模块,包括RMSD、RMSF的计算 |
| MD Hbond | 轨迹氢键分析工具 |
| MD Distance | 轨迹距离分析工具,输出指定原子、残基之间动态距离变化 |
| MD Clustering | 轨迹聚类分析工具 |
| MD PCA | 轨迹主成分分析工具 |
| MD Gyration | 回旋半径分析工具 |
| MD SASA | 计算指定组别的溶剂可及表面积 |
Trajectory analysis involves structural analysis of the trajectories generated from molecular dynamics simulations to observe the dynamic changes of the study object during the simulation process.
| Module/Workflow Name | Description |
|---|---|
| MD Trajectory | Extracts trajectories from equilibrium simulations based on start frame, end frame, and interval frame, and converts them to GRO or PDB format files |
| MD RMS | System structure stability analysis module, including calculations of RMSD and RMSF |
| MD Hbond | Trajectory hydrogen bond analysis tool |
| MD Distance | Trajectory distance analysis tool, outputs dynamic distance changes between specified atoms or residues |
| MD Clustering | Trajectory clustering analysis tool |
| MD PCA | Trajectory principal component analysis tool |
| MD Gyration | Radius of gyration analysis tool |
| MD SASA | Calculates the solvent accessible surface area of specified groups |
结合自由能计算是预测分子间结合强弱的重要方法。
| 模块/流程名称 | 描述 |
|---|---|
| MMPBSA | 计算受体与配体之间的结合自由能,并且提供能量分解数据等数据 |
| Alanine Scan (MMPBSA) | 计算丙氨酸突变后的结合自由能,并且提供能量分解数据 |
| MMPBSA of One Protein/DNA Structure | 计算一帧蛋白-蛋白复合物/蛋白-核酸复合物结构的结合自由能流程 |
| MMPBSA of One Protein-Ligand Structure | 计算一帧蛋白-小分子结构的结合自由能流程 |
| PPI Binding Energy (Graphomer) | 蛋白-蛋白复合物结合能模块,基于图transformer模型预测蛋白-蛋白结合亲和力 |
| PPI Binding Energy & Contacts | 蛋白-蛋白复合物结合能与相互作用分析模块,基于界面接触特征预测蛋白-蛋白结合亲和力 |
Combining free energy calculations is a crucial method for predicting the strength of molecular interactions.
| Module/Workflow Name | Description |
|---|---|
| MMPBSA | Calculates the binding free energy between receptor and ligand, and provides energy decomposition data |
| Alanine Scan (MMPBSA) | Calculates the binding free energy after alanine mutation, and provides energy decomposition data |
| MMPBSA of One Protein/DNA Structure | Workflow for calculating the binding free energy of a single protein-protein or protein-nucleic acid complex structure |
| MMPBSA of One Protein-Ligand Structure | Workflow for calculating the binding free energy of a single protein-small molecule structure |